Microbial Reference Genomes
The HMP sequenced over 2000 reference genomes isolated from human body sites, collected from publicly available sources. The information gained from the reference genomes aids in taxonomic assignment and functional annotation of 16S rRNA and metagenomic wgs sequence, respectively, from microbiome samples.
About Reference Genomes
The HMP's intial aim was to sequence 3000 genomes from both cultured and uncultured bacteria, plus several viral and small eukaryotic microbes isolated from human body sites. This, in conjunction with reference genomes sequenced by HMP Demonstration Projects and other members of the International Human Microbiome Consortium (IHMC), were to supplement the available selection of non-HMP funded human-associated reference genomes to provide a comprehensive pool of genome sequences to aid in the analysis of human metagenomic data.
Sequencing efforts were largely bacterial-focused, however a smaller number of viruses, archaea and small eukaryotic references were included. Reference genomes were selected from the same major body sites as HMP metagenomic sampling efforts, as well as additional body sites not targeted by HMP metagenomic sampling, to provide the most thorough representation.
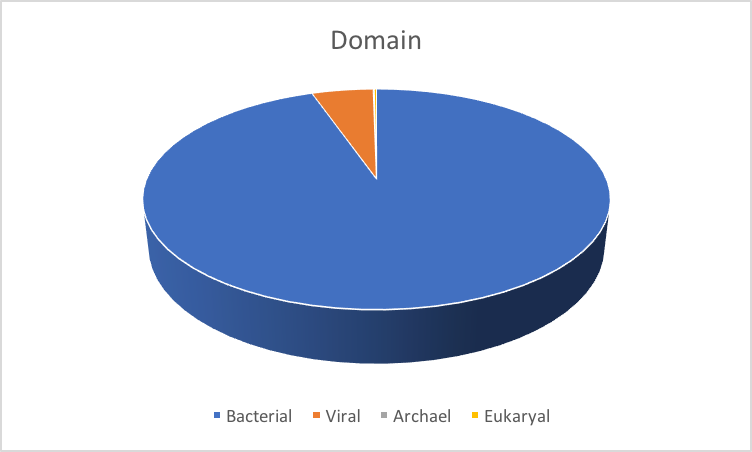
The majority of the Microbial Reference Genomes were sequenced only to a high-quality draft stage. High-quality draft sequences do not include every base of the genome, rather they are assemblies of several large contiguous pieces of sequence (contigs) with subsequent gaps in sequence knowledge. Nonetheless, they provide enough information for a general assessment of gene content. About 15% of the Microbial Reference Genomes were taken to improved levels of finishing up to and including a finished (often referred to as Gold Standard) genome.
The community-defined genome finishing standards adopted by the HMP were aggregated from the following references:
- P. S. G. Chain, et al., Genome Project Standards in a New Era of Sequencing. Science 9 October 2009: Vol. 326. no. 5950, pp. 236 - 237
- The Human Microbiome Jumpstart
Reference Strains Consortium, A Catalog of Reference Genomes from the
Human Microbiome.
Science 21 May 2010:Vol. 328. no. 5981, pp. 994 - 999
Metadata pertaining to all reference genomes, including body site, finishing status and links to NCBI and public repositories, can be found in the HMP Project Catalog. Sequence data is available through the legacy DACC Data Browser, or through NCBI BioProject 28331.
Reference genome efforts were coordinated through the HMP Strains Working Group. This working group included representatives of the HMP Sequencing Centers, DACC, Technology Development grantees, and a group of body site experts providing feedback and recommendations on strain selection. Reference genomes were collected from public repositories, through collaborations with sequencing centers, and via community feedback.
Analyses describing the initial set of HMP reference genomes were published in 2010:
- Human Microbiome Jumpstart Reference Strains Consortium., A catalog of reference genomes from the human microbiome. Science 21 May 2010: 328(5981), pp. 994-97.
Why Sequence Reference Genomes?
Reference genomes serve as guideposts to aid metagenomic analysis.
Within the human body, it is estimated that there are 10x as many microbial cells as human cells. Our microbial partners carry out a number of metabolic reactions that are not encoded in the human genome and are necessary for human health. Therefore when we talk about the "human genome" we should think of it as an amalgam of human genes and those of our microbes.
The majority of microbial species present in the human body have never been isolated, cultured or sequenced, typically due to the inability to reproduce necessary growth conditions in the lab. Therefore there are huge amounts of organismal and functional novelty still to be discovered. Metagenomics, or the study of microbial communities, will, in theory, reveal this novel content. In order to assign metagenomic sequence to taxonomic and functional groupings, and to differentiate the novel from the previously described, it is necessary to have a large pool of described genomes (aka reference genomes) from the same environments. Reference genomes are used to map taxonomic and functional information onto metagenomic sequence.
The HMP sequenced over 2000 microbial genomes, however we are still far from covering the breadth of phylogenetic diversity of the human microbiome. The majority of microbes present in the body are currently uncultured using standard methods. Recent advances in culture and single cell based technologies are making access to these previously inaccessible microbes possible. The HMP Strains Working Group worked closely with technology development grantees using novel culture- and single cell-based techniques, to sequence previously uncultured and rare species found in human microbiome samples. Using methods incorporating 16S-based surveys of metagenomic samples, the HMP Working Group compiled a list of as of yet unsequenced members of the microbiome, prioritized based on their distance from already sequenced genomes and their frequency among samples. See Most Wanted Taxa for more information.
HMP Project Catalog
The HMP Project Catalog provides metadata for all human associated isolate reference genomes. Metadata collected for sequencing projects complies with the Genomic Standards Consortium MIGS minimum information requirements. The Catalog is built upon the Genomes OnLine (GOLD) database structure and the IMG-GOLD system for capturing genome project information.
Users of this resource can:
- View all HMP/IHMC reference genomes, along with links to data and resource repositories
- Search for a specific species or strain(s) of interest
- Filter on over 60 metadata fields, including body site, taxonomic group, project status, sequencing center, and more
- Show and hide columns to generate custom tables
- Export custom tables as excel or csv files
View Entire Project Catalog
Access to Strains
Early in the project, NIH released the following statement on repository deposition and center sequencing:
The HMP Consortium has made a commitment to making available bacterial strains whose genomes are sequenced as part of the HMP. Many of these strains are already available in public repositories, such as ATCC and DSMZ, but for those strains not available, the HMP will ensure the deposition of strains into the Biodefense and Emerging Infections Research Resources Repository (BEI). The deposition process includes a number of steps required by both the collaborator making the deposit and BEI. In order to prevent unnecessary delays in sequencing, the HMP sequencing centers and NIH staff have agreed to a specific time point at which sequencing can be initiated. Sequencing can begin when the collaborator depositing this strain has contacted BEI and has submitted the deposit forms to BEI. This corresponds with the deposition status of "BEI Contacted" on the HMP DACC. Even though this allows sequencing to begin before the strain is fully available at BEI, the HMP maintains its commitment to ensuring each sequence strain becomes publicly available.

BEI, Biodefense and Emerging Infections Research Resources Repository, was established by the National Institute for Allergy and Infectious Diseases (NIAID) and is managed under contract by ATCC, to provide reagents, tools and organisms for research of category A, B and C pathogens and emerging infectious disease agents, and in the case of the Human Microbiome Project, HMP reference genomes to the general research community. Materials requested from BEI are provided at no cost, aside from shipping & handling. Visit BEI for more information about their role in this project. Please note that BEI maintains a limited stock of HMP reference genomes. Once a particular stock is depleted, that strain will no longer be made available through this resource.
Reference genome organisms already available from a public repository are not required to be deposited in BEI. These repositories include ATCC, DSMZ, CCUG, JCM, LCDC and NCTC.
Refer to the Resource Repository column of the
HMP Project Catalog for repository identifiers for individual projects, where available.



