Microbiome Analyses
The HMP performed 16S rRNA and metagenomic sequencing of samples from a healthy human population to address questions such as whether there is a "core" microbiome at individual body sites and whether variation in the microbiome can be systematically studied.
About HMP Metagenomic Sequencing & Analysis
Within the human body, it is estimated that there are 10x as many microbial cells as human cells. Our microbial partners carry out a number of metabolic reactions that are not encoded in the human genome and are necessary for human health. Therefore when we talk about the "human genome" we should think of it as an amalgam of human genes and those of our microbes.
The majority of microbial species present in the human body have never been isolated, cultured or sequenced, typically due to the inability to reproduce necessary growth conditions in the lab. Therefore there are huge amounts of organismal and functional novelty still to be discovered. Metagenomics, or the study of microbial communities, takes advantage of advances in sequencing technology and analysis methods to comprehensively examine microbial communities directly from their natural habitats, potentially revealing novel content. The HMP used 16S rRNA and whole metagenome shotgun (mWGS) sequencing to characterize the complexity of the human microbiome at 15 or 18 body sites. This supplements the sequencing and analysis of reference genomes isolated from human body sites, generating unprecedented amounts of data about the complexity of the human microbiome, and providing a baseline for further research into the impacts of the microbiome on human health and disease.
16S and mWGS sequencing occurred in a four-phase process, consisting of a mock data pilot phase, a clinical pilot phase, clinical production phase 1 completed in July 2010, and clinical production phase 2 completed in 2013. The following diagram shows the sample flow of the clinical phases of the project.
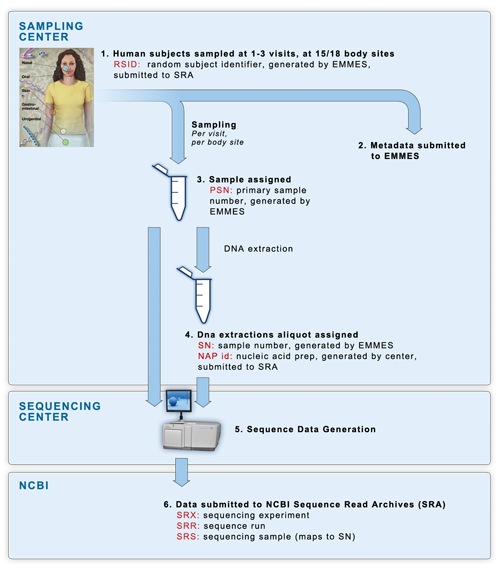
(1) Samples were collected at 2 dedicated
sampling centers, with each human
participant being sampled at 15 or 18 body sites over one to three visits.
(2)
All subject & sample metadata was submitted to EMMES, our partner in
metadata storage and management. EMMES assigned each human subject a
de-identified RSID (random subject identifier).
(3) Every individual sample was assigned a PSN id, an EMMES-generated
Primary Sample Number.
(4) DNA was extracted from the primary samples using
methods described in the
Manual of
Procedures, and assigned a NAP id (Nucleic Acid Preparation) generated by
the sequencing centers. Extractions were divided into aliquots as necessary.
Each aliquot was assigned an SN id, an EMMES-generated Sample Number. For the
clinical pilot phase, every primary sample was sent to two sequencing centers
to assess reproducibility of technical replicates across multiple institutions.
Therefore there is a one-to-many relationship between PSN and SN ids. In the
non-pilot clinical phases one and two, a single PSN sample generated a single SN sample, which
was sent to a single sequencing center, allowing for a one-to-one relationship
between PSN and SN.
(5) Sequencing centers performed sequencing on each SN
sample, sequencing 16S variable regions 3-5 (V35) from every samples within the
study, and variable regions 1-3 (V13) and variable regions 6-9 (V69) from
subsets of samples. If the SN samples did not provide enough material for
sequencing, the centers were sent the original PSN samples for re-extraction
and sequencing.
(6) All sequence data was submitted by the sequencing centers directly to
NBCI's Sequence Read Archives (SRA) data repository. Centers submitted data
using the RSID subject ids, and the NAP sample ids. Data was then organized at
NCBI into their standardized levels of experiment accession (SRX ids), run id
(SRR ids) and sample id (SRS ids).
Public metadata associated with mWGS samples, limited to Subject ID, gender, visit number, body site can be found in the HMP Project Catalog. Sequence data is available through the HMP Portal, the legacy DACC Data Browser, or through NCBI BioProject 48489 (16S) and NCBI BioProject 43017 (mWGS). Tools and protocols used and/or developed by the HMP consortium are available at Tools and Technology.
Metagenomic sequencing and analysis efforts were coordinated through a large number of working groups covering all aspects from sequence generation & processing, through data release. These working groups included representatives of the HMP Sequencing Centers, DACC, Technology Development grantees, and a group of body site experts.
Sample Collection & Participant Guidelines
16S rRNA and metagenomic sequencing was performed on samples collected from 300 healthy adult men and women between the ages of 18 and 40, recruited at Baylor College of Medicine in Houston, TX and Washington University in St. Louis, MO. See a full list of study inclusion/exclusion criteria, as well as the consent forms and other information provided to participants by the two universities:
- Baylor College of Medicine Consent Form
- Baylor College of Medicine Patient Information
- Washington University Consent Form
- Washington University Patient Information
- Washington University Protocol Amendment
Samples were collected in a non-invasive manner from five major body sites, with a total of 15 or 18 specific body sites. In addition, subjects donated blood and serum for possible future use examining the relationship of host genotype to an individual's microbiota, and determining immune re\ sponse to organisms identified in the individual's microbiome, respectively. All enrolled subjects were sampled at one visit, with a subset of subject sampled at up to three visits. Patient screening began in November 2008 and the final sample was\ taken in October 2010, for a total of over 11,000 primary specimens from 300 adults.
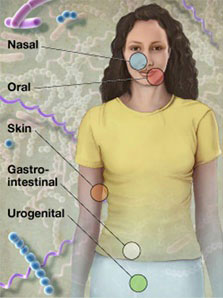
- Oral cavity
- Attached keratinized gingiva (gums)
- Buccal mucosa (cheek)
- Hard palate
- Palatine tonsils
- Saliva
- Subgingival plaque
- Supragingival plaque
- Throat
- Tongue dorsum
- Nasal cavity
- Anterior nares (nostrils)
- Skin
- Left and right antecubital fossa (inner elbow)
- Left and right retroauricular crease (behind the ear)
- Gastrointestinal tract
- Stool
- Urogenital tract
- Mid vagina
- Posterior fornix
- Vaginal introitus
See the HMP Manual of Procedures - Core Microbiome Sampling Protocol for more information on sampling procedures used for collecting metagenomic samples under the HMP.
Former study participants who have questions or concerns about any aspect of the project should contact:
Baylor College of Medicine
James Versalovic, MD, PhD
Baylor College of Medicine
Texas Children's Hospital
6621 Fannin Street, MC 1-2261
Houston, TX 77030
phone: (832) 824-2213
email: jamesv@bcm.edu
Wendy Keitel, MD
Department of Molecular Virology and Microbiology
Baylor College of Medicine 280
One Baylor Plaza
Houston, TX 77030
phone: (713) 798-5250
email: wkeitel@bcm.edu
Washington University
Mark Watson, MD, PhD
Washington University School of Medicine
Campus Box 8118
660 S. Euclid Avenue
St. Louis, MO 63110
phone: (314) 454-7919
email: watsonm@wustl.edu
Michael Dunne, PhD
Washington University School of Medicine
258A Barnes-Jewish Hospital Service Building
St. Louis, MO 63110
phone: (314) 362-1547
email: dunne@wustl.edu
Validation using mock communities
At the start of the HMP, there was no standardized protocol for ensuring high throughput consistency of 16S amplification & sequencing protocols. Therefore the HMP evaluated a number of protocols using a synthetic mock community of 21 known organisms, before adopting the HMP 16S 454 Protocol. These organisms include a variety of genera commonly found on or within the human body. Genomic DNA from each organism was mixed, based on qPCR of 16S rRNA measurements, to generate two mock mixtures: a) an even mock community, containing 100,000 16S copies per organism per aliquot, and b) a staggered mock community, containing 1,000 to 1,000,000 16S copies per organism per aliquot
See mock community composition here
Mock communities were also used to evaluate consistency between sequencing centers of both 16S rRNA and metagenomic wgs sequencing.
Mock communities are available to the community through the BEI Resource.

BEI, Biodefense and Emerging Infections Research Resources Repository, was established by the National Institute for Allergy and Infectious Diseases (NIAID) and is managed under contract by ATCC, to provide reagents, tools and organisms for research of category A, B and C pathogens and emerging infectious disease agents, and in the case of the Human Microbiome Project, HMP reference genomes and mock community microbiome samples to the general research community. Materials requested from BEI are provided at no cost, aside from shipping & handling. Please note that BEI maintains a limited stock of HMP mock community samples. Once a stock is depleted, that sample will no longer be available through this resource.
- Cell mixture
- Stored in PBS (BEI:HM-280)
- Stored in PBS + 40% glycerol (BEI:HM-281)
- Genomic DNA extract
- Even community (BEI:HM-278D)
- Staggered community (BEI:HM-279D)
Mock community sequence data can be found at NCBI BioProject 48475. The DACC hosts a summary table of community composition at HMMC.
16S Sequencing & Analysis
16S rRNA sequencing has been used to characterize the complexity of microbial communities at each body sites, and to determine whether there is a core microbiome.
The 16S rRNA sequence contains both highly conserved and variable regions. These variable regions, nine in number (V1 through V9), are routinely used to classify organisms according to phylogeny, making 16S rRNA sequencing particularly useful in metagenomics to help identify taxonomic groups present in a sample. More about 16S rRNA.
Fundamental to the use of 16S rRNA sequencing and analysis by the HMP was the development of a consistent 16S sequencing protocol used across all groups contributing to this project.
- 16S rRNA Sequence Highlights
- sequencing was performed using the Roche-454 FLX Titanium platform
- V3-V5 variable region window (V35) was sequenced for all samples
- V1-V3 variable region window (V13) was sequenced for a subset of ~3,000 samples, and V6-V9 variable region window (V69) was sequenced for a subset of 100 samples in order to provide a complementary taxonomic view
Raw and trimmed sequence data is available via through the HMP Portal, the legacy DACC Data Browser, or through NCBI BioProject 48489. 16S tools and protocols used and/or developed by the HMP consortium are available at Tools and Technology. DACC members worked closely with the Genome Standards Consortium to ensure that HMP 16S rRNA sample and sequence preparation data was MIMARKS compliant.
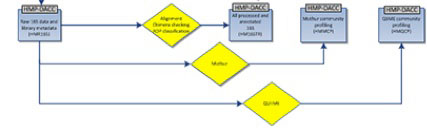
The HMP 16S analysis flow is shown above, with processes as diamonds and data types as squares. This represents a small portion of the HMP Data Flow Diagram published in 2012, and therefore dcoes not necessarily represent more recent analyses. All trimmed 16S samples were processed using Qiime, and the first set of 5,700 samples, frequently referred to as clinical production phase 1, were processed using mothur. Analysis protocols, with software versions, are available at Tools and Technology.
Metagenomic WGS Sequencing and Analysis
Metagenomic whole genome shotgun (mWGS) sequencing provides insights into the functions and pathways present in the human microbiome. HMP data serves as a reference framework for those looking into associations between changes in the human microbiome and disease states.
Unlike 16S rRNA sequencing, mWGS does not target specific regions of the metagenomic sample, rather sequences everything, with more abundant organisms sequenced more deeply than low abundance members of the community. This may include sequencing of species specific, or even strain specific, marker genes, allowing for a finer level of classification of the organisms in a community. Importantly, mWGS provides a picture of the functional potential of the community.

- Metagenomic Shotgun Sequence Highlights
- sequencing was performed using the Illumina GAIIx platform with 101bp paired-end reads
- all samples were screened for human contamination using NCBI's BMTagger tool, with ~49% of reads targeted for removal as human. These reads are subjected to authorized access only, through NCBI dbGAP
- samples underwent quality control assessments, including identification of outliers by mean contig and ORF density, human hits, rRNA hits and size
- samples passing QC were assembled initially using an optimized SOAPdenovo protocol. Samples were later reassembled using IDBA-UD
Raw and assembled sequence data is available via through the HMP Portal, the legacy DACC Data Browser, or through NCBI B\ ioProject 43017. mWGS tools and protocols used and/or developed by the HMP consortium are available at Tools and Technology. Public metadata associated with mWGS samples, limited to Subject ID, gender, visit number, body site can be found in the HMP Project Catalog. DACC members worked closely wit\ h the Genome Standards Consortium to ensure that HMP mWGS sample and sequence preparation data was MIGS compliant.
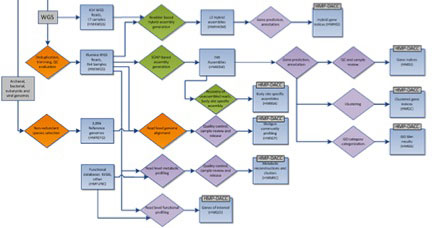
The HMP mWGS analysis flow is shown above, with processes as diamonds and data types as squares. This represents a small portion of the HMP Data Flow Diagram published in 2012, and therefore does not represent more recent analyses. All mWGS assemblies underwent community and functional profiling. Community profiling was done initially by mapping reads to a database of HMP reference genomes, and subsequently via Metaphlan and Metaphlan2, developed during the course of the HMP project. Functional profiling was done using HUMAnN (HMP Unified Metabolic Analysis Network), developed within the HMP, and HUMAnN2, providing gene and pathway level coverage and abundance. In addition, assemblies were annotated to create a gene index and clustered gene index. Analysis protocols, with software versions, are available at Tools and Technology.
Joint 16S/mWGS Analysis
Pairing up 16S and mwGS sequence from a single or set of samples, can initially seem confusing. The following tables join 16S dataset "SN" and "PSN" identifiers with metagenomic dataset "SRS" identifiers. Tables are separated by 16S variable region (V).
HMP Project Catalog
The HMP Project Catalog provides publicly available metadata fields, limited to Subject ID, gender, visit number, body site, from HMP samples sequenced by mWGS. Public metadata collected for sequencing projects complies with the Genomic Standards Consortium MIGS minimum information requirements. To protect subject privacy, additional metadata is available to qualified researchers only through NCBI's dbGaP portal. "Qualified researchers" are defined as PI-level investigators at legitimate institutions wh\ o can describe how they plan to use the data and can follow a series of precautions to safeguard patient privacy. Detailed information on accessing private data is available at the NCBI dbGaP site.
The Catalog is built upon the IMG-GOLD system for capturing metagenomic project information.
Users of this resource can:
- View all HMP mWGS samples reference genomes, along with links to data and resource repositories
- Search for a specific subjects or samples of interest
- Filter on available metadata fields
- Show and hide columns to generate custom tables
- Export custom tables as excel or csv files
View Entire Project Catalog



